
Sommaire
Toutes les cellules de notre corps contiennent la même séquence d’ADN, le même génome. Et pourtant il existe une grande variété de types cellulaires, par exemple les fibres musculaires, les cellules de la peau, du sang ou encore les neurones.
Dans chacun de ces types cellulaires, certains gènes sont exprimés, c’est-à-dire que la séquence d’ADN correspondante est transformée en ARN puis en protéines, alors que d’autres sont éteints. Les instructions qui garantissent une expression coordonnée de ces gènes au cours du développement, puis dans chaque tissu de chaque organe, sont elles-mêmes inscrites dans le génome.
Seuls 2 % de notre séquence d’ADN code pour des protéines et c’est dans les 98 % restant du génome que l’on cherche actuellement à comprendre le programme de coordination de l’expression des gènes.
C’est donc dans un livre de 3 milliards de lettres (imaginez un roman d’un million de pages !) qu’il faut décrypter les règles de ce programme.
C’est là que l’intelligence artificielle va jouer un rôle important. Mais avant de comprendre comment, il nous faut résumer ce que nous savons sur la façon dont est mis en œuvre ce programme. Des coffres verrouillés Les premières réponses obtenues montrent que c’est la manière dont se replie la molécule d’ADN au sein des chromosomes qui définit le programme de régulation des gènes. Une image simplifiée de ce repliement serait celle d’une chambre forte dans une banque.
Chaque coffre contient un gène, et la séquence de ce gène est utilisée pour fabriquer une protéine seulement si ce coffre est ouvert. Chaque coffre est verrouillé par une serrure à combinaison et seules quelques combinaisons sont capables de l’ouvrir.
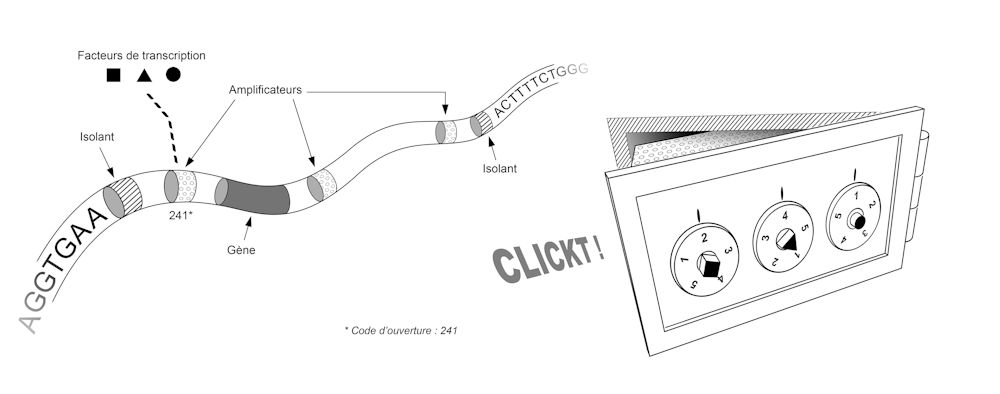
Dans cette image, chaque cellule est une réplique de la chambre forte, une combinaison est un ensemble de facteurs de transcription, et pour chaque gène – chaque coffre – existe une combinaison unique, spécifique à la cellule. Cette combinaison correspond à l’ensemble des facteurs de transcription présents dans la cellule. Ces facteurs de transcription activent ou inhibent les gènes en se liant sur la molécule d’ADN à des endroits précis.
Ainsi, à une combinaison de facteurs de transcription donnée correspond un ensemble de coffres déverrouillés et un ensemble de gènes exprimés. Dans les différentes régions d’un embryon en train de se développer, ces facteurs peuvent être présents ou absents, et la réaction de notre génome à leur présence ou à leur absence permet l’apparition de tissus spécialisés aux endroits voulus. À gauche, un domaine génique contenant un gène, trois amplificateurs et flanqué de deux isolants. Trois facteurs de transcription, représentés par une carrée, un rond, et un triangle, vont, lorsqu’ils sont présents dans la cellule en nombre suffisant, se lier à un amplificateur du gène pour permettre son expression. À droite, chaque domaine génique peut être comparé a un coffre-fort. Dans cette image, les isolants correspondent aux parois du coffre, les amplificateurs aux mécanismes de serrure. Le coffre s’ouvre si et seulement si les bons facteurs de transcription sont présents dans les proportions requises. Jean-François Dejouannet - MNHN / UMS 2AD / AIS, Author provided La séquence de notre génome contient ainsi le plan qui permet de construire non seulement l’ensemble des coffres, mais aussi l’ensemble des systèmes de verrouillage de ces coffres. Ce plan fait intervenir deux types d’éléments, qui correspondent chacun à de petites séquences de quelques dizaines de lettres (A, C, T ou G). Tout d’abord les « isolants » permettent partitionner le génome, c’est-à-dire de définir là où commence et où finit chaque coffre. Ensuite les « amplificateurs » permettent de fabriquer le système de verrouillage qui valide ou invalide l’ouverture de chaque coffre. Pour avoir une idée de la taille et de la complexité de ce système, il faut imaginer que notre génome contient environ 30 000 gènes, un nombre similaire d’isolants, et des dizaines voire des centaines d’amplificateurs pour chaque gène. Des expériences pour mieux comprendre Pour mieux comprendre l’activité de tous ces composants au cours du développement, des expériences de grande ampleur sont réalisées aujourd’hui. Ces expériences reposent sur notre capacité à lire, ou séquencer, la succession des lettres du génome.
Les techniques de séquençage, qui était au paravent seulement utilisable sur des ensembles de millions de cellules, peuvent maintenant être appliquées aux cellules uniques.
Ces développements font entrevoir pour la première fois la possibilité de révéler simultanément l’ensemble des éléments régulateurs (isolants et amplificateurs) ainsi que leur activité dans les différentes cellules au cours du temps.
Bien que les recherches dans ce domaine avancent à grands pas grâce aux nouvelles technologies, une question reste en suspens : comment déterminer l’effet d’une variation du génome sur le processus de régulation des gènes ?
Cette question est d’une importance cruciale pour comprendre pourquoi certaines maladies ont une prédisposition génétique et ainsi comment mieux soigner un individu lorsque l’on connaît son patrimoine génétique.
On observe en effet couramment que certaines variations récurrentes du génome peuvent avoir un rôle dans l’apparition ou l’aggravation de maladies. La grande majorité de ces variations apparaît dans des régions du génome qui ne sont pas des gènes, mais des régions isolantes ou amplificatrices. Un algorithme pour analyser les séquences d’ADN Pour comprendre l’effet de ces variations du génome, il est maintenant possible de recourir à l’intelligence artificielle. L’idée est simple : utiliser toutes les données expérimentales obtenues jusqu’ici pour entraîner un algorithme à prédire l’activité des régions isolantes et amplificatrices en fonction de leur séquence génomique.
Pour cela, il faut tout d’abord convertir les quatre lettres A, C, T et G en langage binaire de 0 et de 1. Puis on entraîne des réseaux de neurones similaires à ceux qu’utilisent les algorithmes de reconnaissance d’images, utilisés par exemple pour numériser des documents manuscrits ou pour analyser les images des caméras embarquées de véhicules autonomes.
Toutes ces applications sont basées sur le même principe : convertir un ensemble de chiffres, appelé entrée, en un autre ensemble de chiffres, appelé sortie. Cette conversion s’obtient en plusieurs étapes. Un ensemble de pré-sorties sont calculées par la multiplication de chaque chiffre de l’entrée par un coefficient puis par l’addition des résultats obtenus. Ce processus est répété en changeant les coefficients pour générer des centaines ou des milliers de pré-sorties qui vont constituer une couche du réseau. L’ensemble des pré-sorties de cette première couche sert d’entrée à une deuxième couche. Plusieurs couches sont ainsi empilées jusqu’à la dernière, qui donne la sortie du réseau. Le processus d’entraînement consiste à fixer les valeurs des coefficients qui font correspondre chacune des entrées à la sortie correspondante. Pour tester ces valeurs et les optimiser, il faut ainsi faire des milliards d’opérations, toutes très simples. Cela est aujourd’hui possible grâce aux performances des cartes graphiques modernes développées initialement pour les jeux vidéo. Un réseau de neurones est entraîné pour associer à une séquence d’ADN sa fonction d’isolant ou d’amplificateur, ainsi que la combinaison correspondant à son activation s’il s’agit d’un amplificateur. Le réseau peut ensuite être utilisé pour scanner le génome en entier et prédire les règles de régulation des gènes d’un individu. Jean-François Dejouannet - MNHN / UMS 2AD / AIS, Author provided Dans notre cas, l’entrée va être une suite de 0 et de 1 qui correspond à une séquence d’ADN binarisée. La sortie va correspondre à une autre suite de 0 et de 1 qui va représenter une annotation fonctionnelle (par exemple 10 ou 01 pour « amplificateur » ou « isolant » et 00001000 ou 01000000 vont correspondre à différentes combinaisons de facteurs de transcription). Une fois l’algorithme entraîné, il devient alors possible de l’utiliser pour prédire l’annotation fonctionnelle d’une séquence dont on aurait changé une ou plusieurs lettres : les fameuses variations. Une équipe de chercheurs de l’Université de Princeton a ainsi testé les variations du génome connues pour être fréquentes chez les personnes autistes et a pu identifier comment elles modifiaient la combinaison de gènes exprimés dans les cellules du cerveau.
La même méthodologie a été appliquée à d’autres maladies parmi lesquelles la maladie de Crohn ou l’infection chronique à l’hépatite B. Dans les prochaines années, la médecine personnalisée devrait pouvoir utiliser cette méthodologie pour adapter un traitement en fonction de données génomiques recueillies pour chaque individu.
Julien Mozziconacci, Professeur en biologie computationelle, Muséum national d’histoire naturelle (MNHN) y Etienne Routhier, Doctorant en data science, Sorbonne Université
Cet article est republié à partir de The Conversation sous licence Creative Commons. Lire l’article original



{kind=link}