Les anticorps, petites protéines produites par le système immunitaire, peuvent se fixer à des parties spécifiques d'un virus pour le neutraliser. Ainsi, pour lutter contre le Covid-19, les laboratoires ont fabriqué des vaccins mais se sont également intéressés aux anticorps synthétiques qui, en se liant aux protéines de pointe du virus, peuvent empêcher le virus de pénétrer dans une cellule humaine. Des chercheurs du MIT ont créé Equidock, un modèle d'apprentissage automatique capable de prédire directement le complexe qui se formera lorsque deux protéines se lieront. La recherche sera présentée à la Conférence internationale sur les représentations de l'apprentissage.
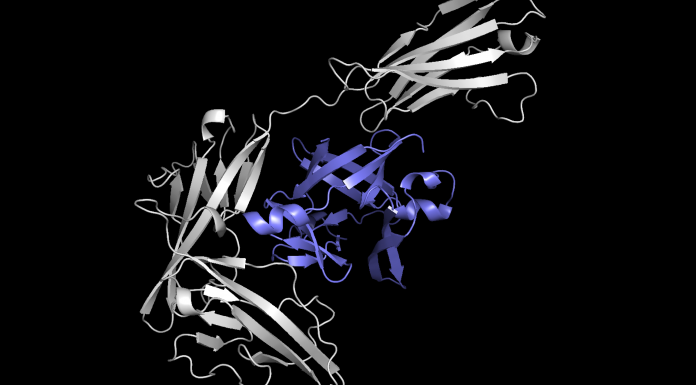
Pour développer un anticorps synthétique réussi, les chercheurs doivent comprendre exactement comment il va s'attacher aux protéines. Ces dernières, avec des structures 3D grumeleuses contenant de nombreux plis, peuvent s'agglutiner dans des millions de combinaisons, donc trouver le bon complexe protéique parmi presque d'innombrables candidats prend énormément de temps.
Octavian-Eugen Ganea, post-doctorant au Laboratoire d'informatique et d'intelligence artificielle du MIT (CSAIL) et Xinyuan Huang, étudiant diplômé à l'ETH Zurich sont les co-auteurs principaux de cette étude. Regina Barzilay, professeur à l'École d'ingénierie pour l'IA et la santé au CSAIL, et Tommi Jaakkola, professeur Thomas Siebel de génie électrique au CSAIL et membre de l'Institut des données, des systèmes et de la société y ont également collaboré.
Equidock, un modèle de deep learning
Pour rationaliser le processus, les chercheurs du MIT ont créé un modèle d'apprentissage automatique capable de prédire directement le complexe qui se formera lorsque deux protéines se lieront. Leur technique est entre 80 et 500 fois plus rapide que les méthodes logicielles de pointe et prédit souvent des structures protéiques plus proches des structures réelles observées expérimentalement. Octavian-Eugen Ganea a déclaré :« L'apprentissage en profondeur est très efficace pour capturer les interactions entre différentes protéines qui sont autrement difficiles à écrire expérimentalement pour les chimistes ou les biologistes. Certaines de ces interactions sont très compliquées et les gens n'ont pas trouvé de bons moyens de les exprimer. Ce modèle d'apprentissage en profondeur peut apprendre ces types d'interactions à partir des données. »
Attachement des protéines
Equidock, se concentre sur l'amarrage du corps rigide, qui se produit lorsque deux protéines se fixent en tournant ou en se déplaçant dans l'espace 3D, mais leurs formes ne se compriment pas ou ne se plient pas. Le modèle prend les structures 3D de deux protéines et convertit ces structures en graphiques 3D qui peuvent être traités par le réseau neuronal. Les protéines sont formées à partir de chaînes d'acides aminés, et chacun de ces acides aminés est représenté par un nœud dans le graphique. Les chercheurs ont intégré des connaissances géométriques dans le modèle, afin qu'il comprenne comment les objets peuvent changer s'ils sont tournés ou déplacés dans l'espace 3D. Le modèle intègre également des connaissances mathématiques qui garantissent que les protéines se fixent toujours de la même manière, quel que soit leur emplacement dans l'espace 3D, comme elles le font dans le corps humain. Grâce à ces informations, Equidock identifie les atomes des deux protéines les plus susceptibles d'interagir et de former des réactions chimiques, appelées points de poche de liaison. Ensuite, il utilise ces points pour placer les deux protéines ensemble dans un complexe. Octavian-Eugen Ganea explique :« Si nous pouvons comprendre à partir des protéines quelles parties individuelles sont susceptibles d'être ces points de poche de liaison, alors cela capturera toutes les informations dont nous avons besoin pour placer les deux protéines ensemble. En supposant que nous puissions trouver ces deux ensembles de points, nous pouvons simplement découvrir comment faire pivoter et traduire les protéines afin qu'un ensemble corresponde à l'autre ensemble. »L'une des plus grandes difficultés de la construction de ce modèle a été le manque de données de formation. Octavian-Eugen Ganea ajoute :
« Parce qu'il existe si peu de données expérimentales 3D pour les protéines, il était particulièrement important d'intégrer les connaissances géométriques dans Equidock, explique Ganea. Sans ces contraintes géométriques, le modèle pourrait détecter de fausses corrélations dans l'ensemble de données. »
Une prédiction quasi-immédiate
Une fois le modèle formé, les chercheurs l'ont comparé à quatre méthodes logicielles. Equidock est capable de prédire le complexe protéique final après seulement une à cinq secondes. Toutes les lignes de base ont pris beaucoup plus de temps, entre 10 minutes et une heure ou plus. Dans les mesures de qualité, qui calculent à quel point le complexe protéique prédit correspond au complexe protéique réel, Equidock était souvent comparable aux lignes de base, mais il les a parfois sous-performés. Octavian-Eugen Ganea précise :« Nous sommes toujours en retard sur l'une des lignes de base. Notre méthode peut encore être améliorée, et elle peut encore être utile. Il pourrait être utilisé dans un très grand criblage virtuel où nous voulons comprendre comment des milliers de protéines peuvent interagir et former des complexes. Notre méthode pourrait être utilisée pour générer très rapidement un ensemble initial de candidats, puis ceux-ci pourraient être affinés avec certaines des méthodes traditionnelles plus précises, mais plus lentes. »En plus d'utiliser cette méthode avec des modèles traditionnels, l'équipe souhaite incorporer des interactions atomiques spécifiques dans Equidock afin qu'il puisse faire des prédictions plus précises. Par exemple, parfois, les atomes des protéines se fixent par des interactions hydrophobes, qui impliquent des molécules d'eau. Cette technique pourrait aider les scientifiques à mieux comprendre certains processus biologiques qui impliquent des interactions protéiques, comme la réplication et la réparation de l'ADN, ce qui pourrait également accélérer le processus de développement de nouveaux médicaments. Octavian-Eugen confirme :
« Notre technique pourrait également être appliquée au développement de petites molécules ressemblant à des médicaments. Ces molécules se lient aux surfaces des protéines de manière spécifique, donc déterminer rapidement comment cette fixation se produit pourrait raccourcir le calendrier de développement de médicament. »À l'avenir, ils prévoient d'améliorer Equidock afin qu'il puisse faire des prédictions pour l'amarrage flexible des protéines. Le plus gros obstacle est le manque de données pour la formation, l'équipe vise à générer des données synthétiques pour améliorer le modèle.