Prédire la structure des protéines est un des défis de la recherche en biologie. Le MIT Technology Review a relayé une annonce de Deepmind concernant son outil d’IA, le programme AlphaFold. La société y déclare avait réussi à prédire la structure de presque toutes les protéines connues des scientifiques et propose à tous l’accès gratuit à sa base de données de plus de 200 millions de protéines. Parallèlement, des chercheurs du MIT ont publié l'étude « Benchmarking AlphaFold-enabled molecular docking predictions for antibiotic discovery » qui souligne que des améliorations seront nécessaires pour tirer pleinement parti des structures protéiques fournies par AlphaFold.
Deepmind a proposé une 1ère version d'AlphaFold, un système d'IA de prédiction de structures des protéines à la CASP (Critical Assessment of protein Structure Prediction) en 2018, il y a obtenu la 1ère place tout comme AlphaFold 2, la seconde version à laquelle nous avions également consacré un article dans notre magazine papier ActuIA N°3, l'a fait en 2020.
Selon Deepmind, AlphaFold, son système d’IA qui prédit la structure 3D d’une protéine à partir de sa séquence d’acides aminés, atteint régulièrement une précision compétitive par rapport à l’expérience.
En 2021, elle a publié l’article scientifique et le code source expliquant comment elle avait créé ce système d'IA et s'est associée à l’Institut Européen de Bioinformatique de l’EMBL (EMBL-EBI) pour créer la plateforme AlphaFold DB afin de rendre ces prédictions librement accessibles à la communauté scientifique. La dernière version de la base de données contient plus de 200 millions d’entrées, offrant une large couverture d’UniProt, le référentiel standard de séquences et d’annotations de protéines.
AlphaFold DB se concentre actuellement sur le cas d’utilisation validé dans CASP14 : prédire la structure d’une seule chaîne protéique avec une séquence naturelle. Deepmind et l'EMBL continueront de mettre cette base de données à jour.
Toutefois, Deepmind concède que le système d'AlphaFold a quelques limites. Des chercheurs du MIT ont cherché à les analyser ainsi que son potentiel.
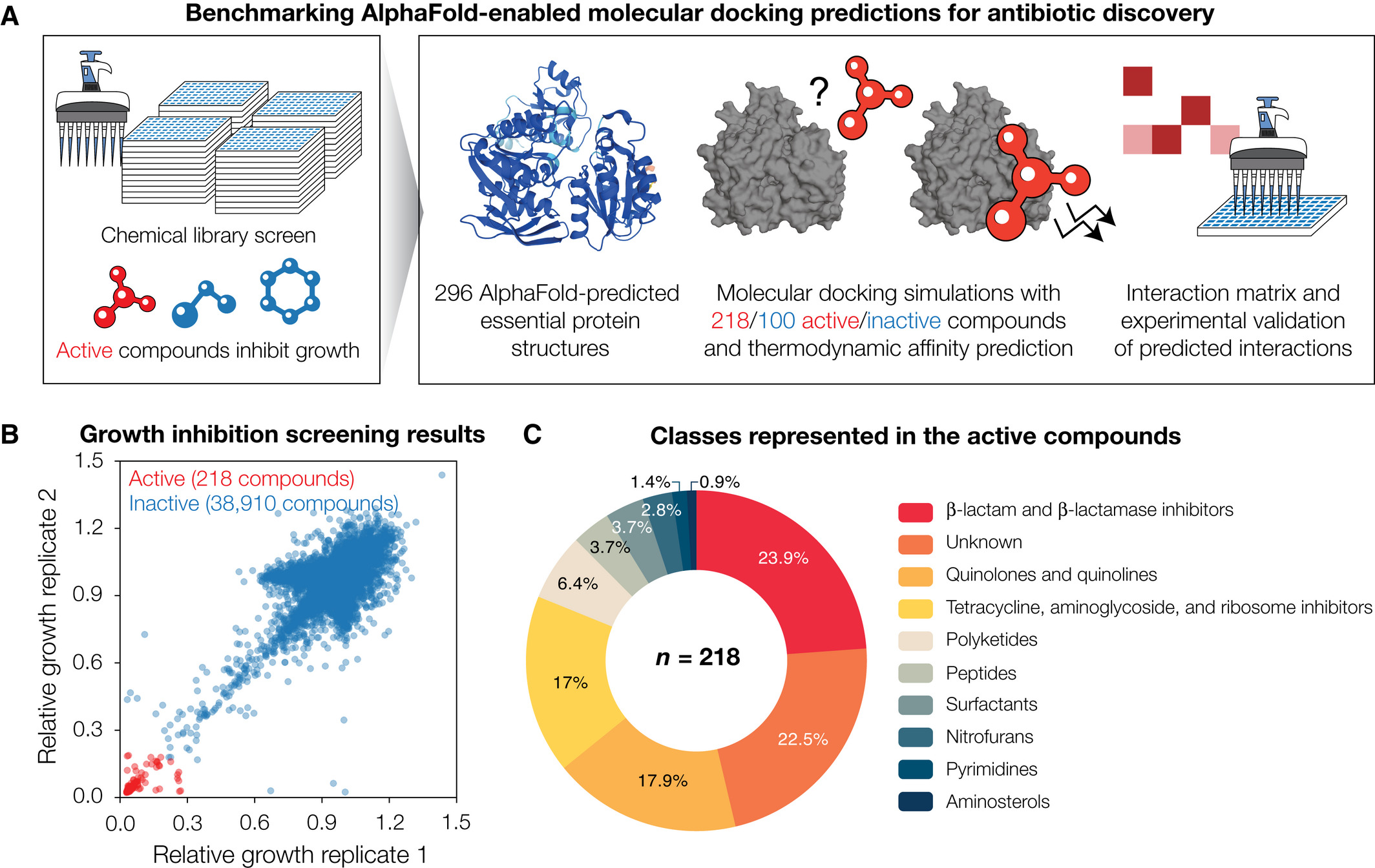 A) Pour définir l'espace chimique d’intérêt, les chercheurs ont effectué des criblages à haut débit de l’inhibition de la croissance contre E. coli de type sauvage. Les composés qui inhibaient la croissance étaient considérés comme actifs, et chaque composé actif était amarré par calcul à chacune des 296 structures protéiques essentielles d’E. coli prédites par AlphaFold2. B) Mesures d’inhibition de la croissance pour 39 128 composés, à partir desquels 218 composés (y compris des antibiotiques connus) ont été identifiés comme actifs contre E. coli BW25113. Les données proviennent de deux réplicats biologiques. Les composés dont la croissance relative moyenne était inférieure à 0,2 ont été classés comme actifs (points rouges) et tous les autres composés ont été classés comme inactifs (points bleus). C) Distribution des classes de composés représentées dans les 218 composés actifs.[/caption]
A) Pour définir l'espace chimique d’intérêt, les chercheurs ont effectué des criblages à haut débit de l’inhibition de la croissance contre E. coli de type sauvage. Les composés qui inhibaient la croissance étaient considérés comme actifs, et chaque composé actif était amarré par calcul à chacune des 296 structures protéiques essentielles d’E. coli prédites par AlphaFold2. B) Mesures d’inhibition de la croissance pour 39 128 composés, à partir desquels 218 composés (y compris des antibiotiques connus) ont été identifiés comme actifs contre E. coli BW25113. Les données proviennent de deux réplicats biologiques. Les composés dont la croissance relative moyenne était inférieure à 0,2 ont été classés comme actifs (points rouges) et tous les autres composés ont été classés comme inactifs (points bleus). C) Distribution des classes de composés représentées dans les 218 composés actifs.[/caption]
L'étude : Analyse comparative des prédictions d’amarrage moléculaire activées par AlphaFold pour la découverte d’antibiotiques
L'équipe de chercheurs, dirigée par James Collins, Professeur Termeer de génie médical et de sciences à l’Institut de Génie Médical et de cience (IMES) et au Département de Génie Biologique du MIT, a cherché à savoir si AlphaFold pouvait prédire avec précision les interactions entre les protéines bactériennes et les composés antibactériens, ce qui pourrait permettre le développement de nouveaux antibiotiques. L’identification efficace des mécanismes d’action des médicaments reste un défi. Les approches d’amarrage computationnel ont été largement utilisées pour prédire les cibles de liaison aux médicaments. Pourtant, de telles approches dépendent des structures protéiques existantes, et des prédictions structurelles précises ne sont disponibles que récemment auprès d’AlphaFold2. Pour cette étude, les chercheurs ont combiné AlphaFold2 avec des simulations d’amarrage moléculaire pour prédire les interactions protéine-ligand entre 296 protéines couvrant le protéome essentiel d’Escherichia coli, et 218 composés antibactériens actifs et 100 composés inactifs, respectivement, indiquant une promiscuité généralisée des composés et des protéines. Ils ont ensuite comparé la performance du modèle en mesurant l’activité enzymatique de 12 protéines essentielles traitées avec chaque composé antibactérien. Cela leur a permis de confirmer une promiscuité étendue, mais aussi de constater que la surface moyenne sous la courbe caractéristique de fonctionnement du récepteur (auROC) est de 0,48, ce qui indique de faibles performances du modèle. Les chercheurs ont démontré que le rescoring des poses d’amarrage à l’aide d’approches basées sur le machine learning améliore les performances du modèle, ce qui se traduit par des auROC d'une moyenne de 0,63. Ils ont également constaté que les ensembles de fonctions de correction améliorent la précision de la prédiction et le rapport entre le taux de vrais positifs et le taux de faux positifs. [caption id="attachment_39364" align="aligncenter" width="2128"] A) Pour définir l'espace chimique d’intérêt, les chercheurs ont effectué des criblages à haut débit de l’inhibition de la croissance contre E. coli de type sauvage. Les composés qui inhibaient la croissance étaient considérés comme actifs, et chaque composé actif était amarré par calcul à chacune des 296 structures protéiques essentielles d’E. coli prédites par AlphaFold2. B) Mesures d’inhibition de la croissance pour 39 128 composés, à partir desquels 218 composés (y compris des antibiotiques connus) ont été identifiés comme actifs contre E. coli BW25113. Les données proviennent de deux réplicats biologiques. Les composés dont la croissance relative moyenne était inférieure à 0,2 ont été classés comme actifs (points rouges) et tous les autres composés ont été classés comme inactifs (points bleus). C) Distribution des classes de composés représentées dans les 218 composés actifs.[/caption]
Les résultats de l'étude
L'étude a démontré le potentiel de l’IA à rationaliser le processus de sélection des futurs antibiotiques. Cependant, les résultats suggèrent que, bien qu’AlphaFold2 puisse fournir des informations structurelles riches, des méthodes permettant de modéliser plus précisément les interactions protéine-ligand sont nécessaires pour mieux exploiter AlphaFold2 pour la découverte de médicaments. Références de l'article : « Benchmarking AlphaFold-enabled molecular docking predictions for antibiotic discovery » doi.org/10.15252/msb.202211081 Publication dans la revue « Molecular Systems Biology » le 6 septembre 2022. Auteurs : Wong F, Krishnan A, Zheng EJ, Stark H, Manson AL, Earl AM, Jaakkola T, Collins JJ. 18 : e11081. Affiliations: Félix Wong1,2,3,Aarti Krishnan1,2,3, Erica J Zheng3,4, Hannes Stärk5, Abigail L Manson3, Ashlee M Earl3, Tommi Jaakkola5 etJames J. Collins 1,2,3,6- 1Institute for Medical Engineering & Science, Massachusetts Institute of Technology, Cambridge, MA, USA
- 2Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA
- 3Infectious Disease and Microbiome Program, Broad Institute of MIT and Harvard, Cambridge, MA, USA
- 4Program in Chemical Biology, Harvard University, Cambridge, MA, USA
- 5Computer Science and Artificial Intelligence Laboratory, Massachusetts Institute of Technology, Cambridge, MA, USA
- 6Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA, USA.